



W niedawnym badaniu opublikowanym w Komunikacja naturynaukowcy opracowali podejście do łączenia informacji genomowych, przedstawionych jako sekwencje wirusowe całego genomu, oraz informacji epidemiologicznych, w celu oszacowania odstępu między sekwencjami (SI), zwłaszcza w przypadkach niewystarczających danych dotyczących śledzenia kontaktów.
W kontroli chorób zakaźnych przerwy w sekwencji są krytyczne, ponieważ wymagają informacji o poszczególnych ekspozycjach i śledzenia kontaktów. Obecne podejścia najlepiej nadają się do małych, ograniczonych grup z dużą liczbą próbek; Jednak szacunki z małych wczesnych ognisk są często wykorzystywane w analizie epidemiologicznej na dużą skalę.
Chociaż genomowe badania epidemiologiczne mogą wpływać na działania w zakresie zdrowia publicznego, ograniczenia budżetowe i obawy dotyczące prywatności ograniczają powszechne zgłaszanie i stosowanie.
o studiowaniu
W tym badaniu naukowcy przedstawiają alternatywną, wydajną strukturę wykorzystującą sekwencje wirusów do oszacowania odstępów między seriami w celu zbadania szacunków SI specyficznych dla klastra w ramach pierwszej i drugiej fali koronawirusa 2019 (COVID-19) w Wiktorii w Australii.
Zespół skupił się na specyficznym dla klastra przewidywaniu MS, kluczowego parametru odzwierciedlającego częstość występowania choroby zakaźnej, który jest opisywany jako czas między wystąpieniem objawów w przypadkach pierwotnych i wtórnych. Aby wywnioskować rozkład SI w niekompletnie pobranych próbkach grup przypadków, wykorzystano sekwencje wirusów zamiast bezpośrednich danych na temat par infekcji.
Niepewność u zakażonych i zakażonych osób została wprowadzona poprzez wybranie realnych, specyficznych sieci transmisyjnych w oparciu o sekwencję wirusową i czas wystąpienia znanych objawów.
Biorąc pod uwagę, że wywnioskowana transmisja może nie być transmisją bezpośrednią, przeprowadzono modelowanie mieszaniny w celu oszacowania SI. Ta technika jest przeznaczona dla większych środowisk o niższym poziomie pobierania próbek i różnorodności genetycznej, w których może nie być wystarczających dowodów, aby z pewnością zrekonstruować wzorce przenoszenia.
Naukowcy zsekwencjonowali pełny genom koronawirusa zespołu ostrej niewydolności oddechowej 2 (SARS-CoV-2) i zebrali początek objawów z Wiktorii w Australii.
Zespół zbadał wpływ wykorzystania szacunków odstępów szeregowych specyficznych dla klastrów na ostateczne oszacowania zależnej od czasu liczby reprodukcyjnej (Rt). Walidację przeprowadzono przy użyciu danych epidemiologicznych podobnych do grypy ze znanym rozkładem SI.
W Victorii klastry seryjne liczone były w klastrach transmisji od pierwszej (od 6 stycznia do 14 kwietnia 2020 r.) i drugiej (od 1 czerwca do 28 października 2020 r.) fali COVID-19. RNA dla SARS-CoV-2 wyizolowano z wymazów z jamy nosowo-gardłowej i oznaczono za pomocą reakcji łańcuchowej polimerazy z odwrotną transkrypcją (RT-PCR).
Rekonstrukcje genetyczne przeprowadzono po zmapowaniu danych sekwencji do oryginalnej sekwencji referencyjnej szczepu SARS-CoV-2 Wuhan-Hu-1.
wyniki
Chociaż między pacjentami nie były wymagane żadne dane kontaktowe, a dane zostały pobrane niekompletnie, szacunki SI COVID-19 były porównywalne z tymi obserwowanymi w szeroko zakrojonych badaniach kontaktowych.
Wyniki są bardziej niejednoznaczne niż wiele wcześniej opublikowanych szacunków, chociaż większość szacunków opierała się na małych populacjach z udokumentowanymi parami kontaktowymi, które nie uwzględniają możliwych zaniżonych zgłoszeń.
Skupiska, które występowały w miejscach związanych z długimi spotkaniami, takich jak hospicjum i opieka zdrowotna, wykazywały większe wartości SI niż grupy, które występowały w miejscach uczęszczanych przez krótsze okresy, takich jak przetwórstwo mięsne czy pakowanie.
Wyniki pokazują, że dane genomowe mogą zapewnić perspektywę transmisji na dużą skalę w wysokiej rozdzielczości, ale zestaw danych śledzenia kontaktów może być kosztowny lub niepraktyczny. Szacunki SI były krótsze dla szkół oraz firm zajmujących się przetwórstwem i pakowaniem mięsa niż dla organizacji opieki zdrowotnej.
Sekwencje wirusowe zapewniły wykonalną strategię wnioskowania o szacunkach specyficznych dla klastrów, chociaż podejście to można zastosować w większych sytuacjach, nawet przy braku dokładnych danych śledzenia kontaktów. Dane dotyczące sekwencji patogenów uzyskane od zakażonych osób nie mogą dostarczyć bezpośrednich danych na temat infekcji i zakażonych osób, ale mogą zapewnić perspektywę transmisji w wysokiej rozdzielczości.
Technika działała dobrze w szacowaniu średniej SI, ale ze wzrostem niepewności wraz ze spadkiem odsetka przypadków. Wyniki odchylenia standardowego SI były identyczne.
Oszacowane podejście nie ogranicza się do przypadków, w których dane genetyczne są wykorzystywane do identyfikacji potencjalnych małżonków. Jeśli podano informacje o połączeniu, można je wykorzystać do skonstruowania zestawu potencjalnych sieci transportowych i oszacowania rozkładu SI.
Strategia okazała się skuteczna w warunkach o niewielkiej zmienności transmisji i była również odpowiednia w ustawieniach z dłuższymi odstępami między sekwencjami, o ile sekwencje były wystarczająco zróżnicowane. Niektóre grupy wykazywały większe lub mniejsze ilości próbek: dane te można wykorzystać do monitorowania potencjalnych epidemii, zwłaszcza jeśli metoda jest zintegrowana z nadzorem genetycznym w czasie rzeczywistym.
Wykorzystanie SI klastra do oszacowania RR Wartości w porównaniu z szacunkami opartymi na literaturze wzrosły RR 2-3 krotnie, zwłaszcza w początkowym okresie epidemii.
Podczas pierwszej fali COVID-19 zsekwencjonowano 1242 próbki od 1075 osób z pozytywnym wynikiem SARS-CoV-2, co stanowi 81% zakażeń SARS-CoV-2 zidentyfikowanych w Wiktorii w tym okresie. W przypadku 10 kohort pierwszej fali 312 z 903 próbek, które przeszły kontrolę jakości, należało do grupy genetycznej z ≥15 przypadkami.
Podczas drugiej fali zsekwencjonowano 15 665 próbek od 14 075 osób, co stanowi 84% wszystkich przypadków znalezionych w Wiktorii. Spośród 5745 przypadków, które przeszły kontrolę jakości, 3875 zidentyfikowano jako część kohorty 15 przypadków.
W głównych grupach średni szacunek SI wahał się od 2,6 do 6,7 dnia. Średni pięciodniowy odstęp szeregowy oszacowano za pomocą oszacowania grupy kontaktowej „wszystkich grup”.
Podsumowując, badanie zwróciło uwagę na nową technikę, która łączy dane genetyczne z danymi epidemiologicznymi w celu analizy transmisji genetycznej. Można go zintegrować z monitorowaniem zdrowia publicznego w czasie rzeczywistym, porównywaniem transmisji SARS-CoV-2 i badaniem genomowo określonych sieci pobierania próbek, zapewniając pośredni system epidemiologii genomicznej.

„Odkrywca. Entuzjasta muzyki. Fan kawy. Specjalista od sieci. Miłośnik zombie.”
More Stories
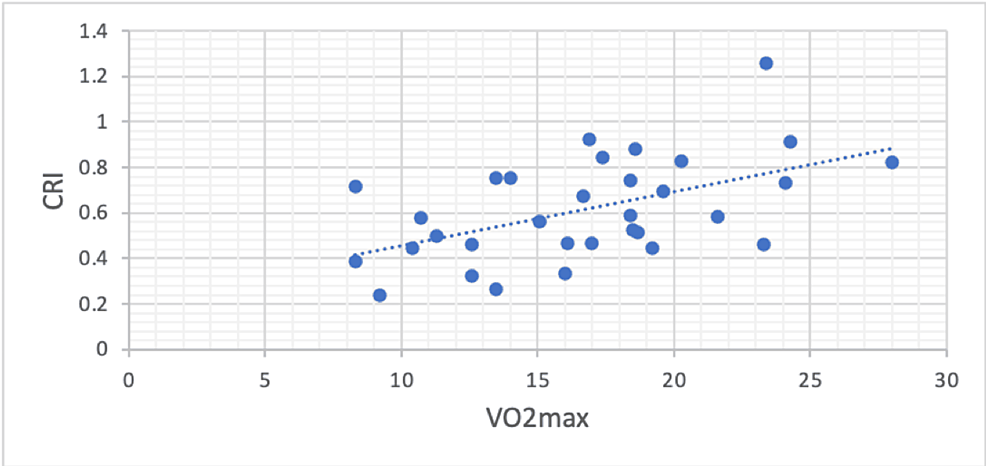
Przywrócenie nieprawidłowej częstości akcji serca i niewydolności chronotropowej podczas ćwiczeń u pacjentów ze śródmiąższową chorobą płuc z nadciśnieniem płucnym lub bez niego.
Badanie wychwytujące ujawnia mutacje BRCA powiązane z gorszymi wynikami w mCRPC
Pielęgniarka z hospicjum staje się popularna, mówiąc o śmierci