


× Zamknąć
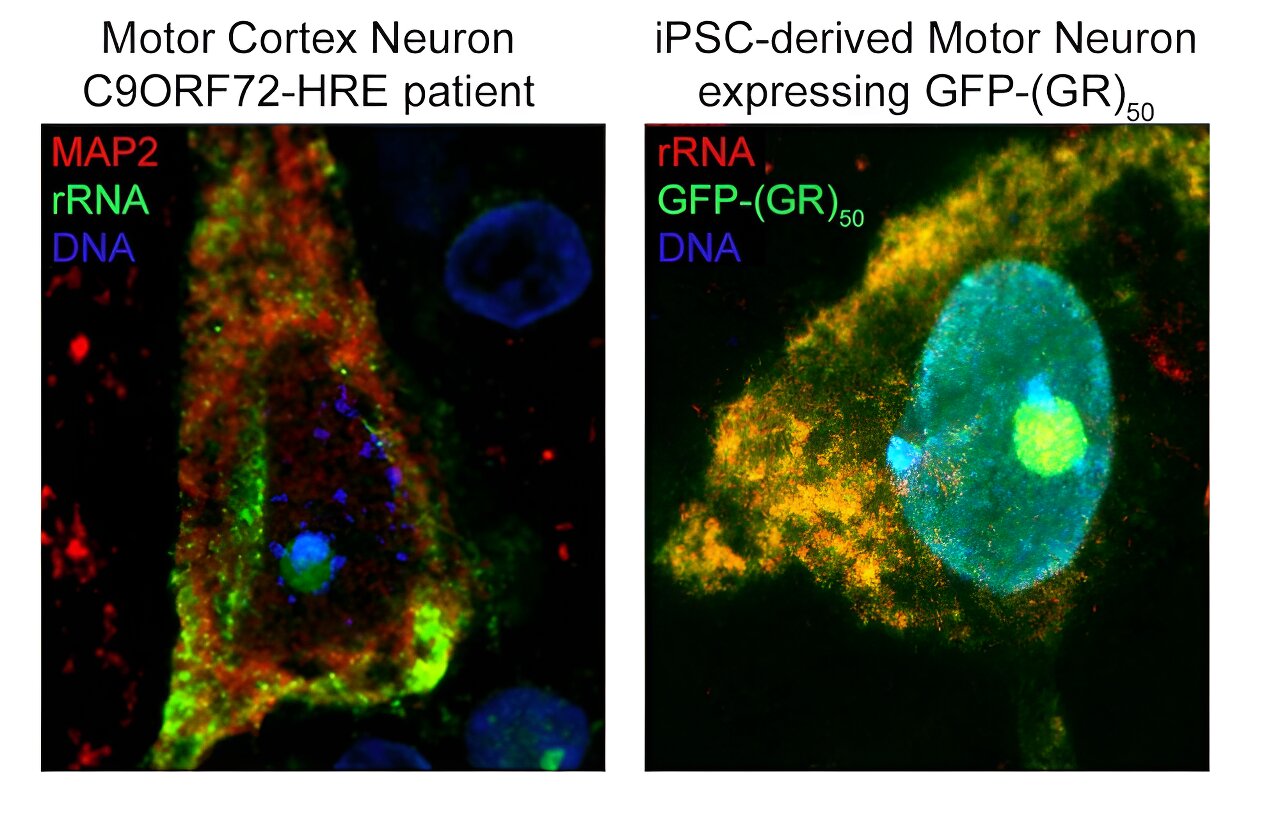
Po lewej: Neuron w korze ruchowej pacjenta z C9ORF72 wybarwiony pod kątem MAP2 i rRNA, pokazujący wzór ekspresji rybosomalnego RNA w ludzkich neuronach, głównie w cytozolu i jądrze. Po prawej: neurony ruchowe pochodzące z iPSC transfekowane lentiwirusem GFP-(GR)50 i wybarwione pod kątem rRNA, pokazujące ogólne nakładanie się poli-GR i rRNA w ludzkich neuronach ruchowych. Źródło: dr Juan Ortega
Dwa badania przeprowadzone w laboratorium dr Evangelosa Keskinisa, profesora nadzwyczajnego na Wydziale Neuronauki Kena i Ruth Davey, Wydział Chorób Nerwowo-Mięśniowych i Neuronauki, ujawniły nowe mechanizmy komórkowe zaangażowane w dwa typy dziedzicznego stwardnienia zanikowego bocznego lub stwardnienia zanikowego bocznego.
Wyniki opublikowane w Postęp nauki I Raporty komórekpoprawiając zrozumienie stwardnienia zanikowego bocznego, postępującej choroby neurodegeneracyjnej atakującej neurony ruchowe w mózgu i rdzeniu kręgowym, a także zapewniając wsparcie dla przyszłego rozwoju terapii celowanych.
„Łapanie” mutacji genetycznych.
Według fundacji Les Turner ALS Foundation w Stanach Zjednoczonych żyje obecnie około 32 000 osób chorych na ALS. Istnieją dwa typy ALS: sporadyczne (niedziedziczne), które stanowi ponad 90% wszystkich przypadków ALS, oraz rodzinne (dziedziczne).
W ich artykule opublikowany W Postęp naukiZespół Keskinisa badał rodzaj rodzinnego ALS spowodowanego powtarzalną sekwencją genetyczną genu C9ORF72, największej genetycznej przyczyny ALS.
Według Keskinisa zdrowe osoby mogą mieć w swoim genomie 20 powtórzeń C9ORF72, podczas gdy pacjenci z ALS mogą mieć ich tysiące. Powtórzenia te ulegają transkrypcji i translacji niekanonicznymi szlakami, które wytwarzają w neuronach nieprawidłowo sfałdowane RNA i dipeptydy (cząsteczki zawierające dwa aminokwasy połączone peptydem).
Zespół Keskinisa postawił hipotezę, że to efekt kaskady prowadzi do toksyczności polegającej na wzmocnieniu funkcji neuronów ruchowych w tego typu stwardnieniu zanikowym bocznym. To skłoniło ich do zbadania mechanizmów, które sprawiają, że te powtarzające się peptydy (R-DPR) stają się coraz bardziej toksyczne.
× Zamknąć
Gdy VCP (po lewej) ulega nadekspresji w kontekście zmutowanego SOD1, zmniejsza się poziom białka SOD1 nierozpuszczalnego w detergentach. Natomiast po chemicznej immobilizacji VCP za pomocą NMS873 (po prawej) następuje znacząca akumulacja białka SOD1 we frakcji nierozpuszczalnej w detergentach. Źródło obrazu: dr Konstantinos Tsiouras
Korzystając z technik obliczeniowych i eksperymentalnych, naukowcy odkryli, że R-DPR silnie wiążą się z cząsteczkami RNA. Następnie zastosowali technikę zwaną usieciowaną immunoprecypitacją, czyli CLIP-seq, w celu wyizolowania określonych fragmentów RNA i odkryli, że R-DPR wiążą się wyłącznie z rybosomalnym RNA. W szczególności dipeptyd poli-GR wiąże się z rybosomalnym RNA, zaburzając homeostazę rybosomów, proces niezbędny do różnicowania komórek i ogólnej struktury komórki.
Wykorzystując te odkrycia, badacze zaprojektowali następnie „przynętę” RNA, która nakłania poli-GR do związania się z czymś, co wydaje się być rybosomalnym RNA – czyli w tym przypadku przynętą. Zarówno w modelach in vivo, jak i w neuronach iPSC pochodzących od pacjentów z mutacjami C9ORF72, cząsteczka przynęty hamowała toksyczność.
„Za pomocą kilku różnych metod pokazaliśmy, że zaprojektowana przez nas cząsteczka – „przynęta” – wiąże się bardzo ściśle i specyficznie z tym białkiem poli-GR, a wiążąc się z nim, zapobiega wiązaniu się go z rybosomalnym RNA i zapobiega przedostawaniu się go do jądra. ” „I tam staje się najbardziej toksyczny” – powiedział Keskinis.
„Służy to jako wstępny dowód na tezę, że cząsteczka RNA posiadająca właściwości „przynęty” może mieć działanie terapeutyczne, i wzmacnia hipotezę, że znaczna część toksyczności wynikającej z mutacji C9ORF72 jest związana z upośledzeniem biologii rybosomów”.
Zdaniem Keskinisa naukowcy pracują obecnie nad udoskonaleniem składu chemicznego cząsteczki przynęty i zamierzają przetestować ją w innych modelach ALS.
„Jesteśmy podekscytowani faktem, że neurony pochodzące z iPSC pacjentów z tymi mutacjami przeżywają dłużej, gdy dajemy im tę cząsteczkę przynęty, co oznacza, że robi coś dobrego. Musimy tylko sprawdzić, czy da się to zrobić „, powiedział Keskinis. terapeutycznie wykonalne i przemieniające”.
Powiązanie genów przyczynowych
Według Keskinisa istnieje 30 znanych genów powiązanych z rodzinnymi postaciami ALS, ale to, czy każdy z genów przyczynia się do tylko jednego ogólnego typu ALS, czy wielu typów, pozostaje wciąż nierozstrzygniętym pytaniem w tej dziedzinie – twierdzi Keskinis.
Aby odpowiedzieć na to pytanie, zgodnie z ich badaniami opublikowany W Raporty komórekzespół Keskinisa wykorzystał indukowane przez pacjenta pluripotencjalne komórki macierzyste (iPSC) neurony ruchowe do opracowania modelu najczęstszego rodzinnego ALS typu 2, który jest spowodowany mutacjami w genie SOD1 i występuje w 2 procentach wszystkich przypadków ALS.
„Z prac wykonanych w tej dziedzinie przez ostatnie 20 lat wiemy, że mutacja ta powoduje toksyczność poprzez efekt wzmocnienia funkcji. Wiemy, że mutacje prowadzą do defektu białka, a defekt rozpoczyna kaskadę prowadzącą do dysfunkcji.” – stwierdził Keskinis. „Ale jak to się dzieje, nie do końca rozumiemy”.
Naukowcy wykorzystali proteomikę czasową do porównania proteomów kontrolnych neuronów iPSC i neuronów iPSC z mutacją SOD1 oraz porównali szybkość degradacji białek (recykling i zastępowanie starych białek) pomiędzy dwiema grupami neuronów.
W neuronach iPSC z mutacją SOD1 odkryli, że inne białko wywołujące ALS, białko zawierające walozynę (VCP), ulegało degradacji wolniej niż neurony iPSC stanowiące kontrolę izogeniczną. Ta powolna degradacja białek ostatecznie doprowadziła do mniejszej interakcji VCP z niektórymi białkami, a większej z innymi.
„To bardzo ekscytujące, ponieważ uważa się, że mutacje w VCP, które mogą powodować ALS, działają w podobny sposób” – powiedział Keskinis.
Następnie badacze starali się ustalić, czy zmieniona funkcja VCP odgrywa rolę w toksyczności zależnej od SDO1. W obu przypadkach w modelach chorych neuronów ze zmutowanymi iPSC SOD1 i C. elegans (rodzaj glisty) zmutowanego SOD1 naukowcy odkryli, że nadekspresja VCP zmniejsza toksyczność, a hamowanie funkcji VCP zwiększa toksyczność.
„Ten artykuł oraz praca, którą wykonaliśmy w naszym laboratorium przez ostatnie kilka lat, pokazują, że chociaż przyczyny genetyczne mogą być różne, zawsze istnieje pewien rodzaj mechanicznego nakładania się, jeśli chodzi o przyczyny dysfunkcji tych komórek” – mówi. powiedział Keyskines.
Jest to ważne, dodał Keskines, ponieważ wyniki mogą pomóc w projektowaniu przyszłych badań klinicznych i udoskonalić stosowanie terapii celowanych. Idąc dalej, Keskinis powiedział, że jego zespół chce ustalić, czy VCP jest skutecznym celem terapeutycznym dla pacjentów z mutacją SOD1, a także innych typów ALS.
więcej informacji:
Juan A. Ortega i wsp., analiza CLIP-Seq umożliwia zaprojektowanie rybosomalnych oligonukleotydów przynętowych RNA chroniących przed wieloma patofizjologami C9ORF72 ALS/FTD, Postęp nauki (2023). doi: 10.1126/sciadv.adf7997
Konstantinos Tsiouras i wsp., Analiza dynamiki degradacji poziomu białek w neuronach pacjentów pochodzących z ALS SOD1 iPSC ujawnia brak równowagi VCP, Raporty komórek (2023). doi: 10.1016/j.celrep.2023.113160

„Odkrywca. Entuzjasta muzyki. Fan kawy. Specjalista od sieci. Miłośnik zombie.”
More Stories
Yokogawa pomaga zrewolucjonizować dziedzinę lipidomiki pojedynczych komórek
WHO obala doktrynę dotyczącą rozprzestrzeniania się chorób przenoszonych drogą powietrzną
Badanie wykazało, że skurcze komórek napędzają początkowe powstawanie ludzkich embrionów